Hydrophobierung von Gips durch Silane (Teil 2)

Über die Wirkungsweise von Silanen als bekannte Hydrophobierungsmittel für Gipsbaustoffe wurde mit experimentellen Untersuchungen am Beispiel von Propyltriethoxysilan im Teil 1 (ZKG 7-8/2013, S. 72-78) berichtet. Im Teil 2 werden mit Hilfe der molekularen Simulation die ablaufenden Vorgänge auf molekularer Ebene im Detail betrachtet und mit den Ergebnissen aus Teil 1 zusammenführend interpretiert, um ein Verständnis über den Wirkungsmechanismus der Hydrophobierung von Gipsbaustoffen durch Silane, speziell der Alkyltriethoxysilane am Beispiel des Propyltriethoxysilans, zu erhalten.
Die Simulationen basierten auf der Annahme, dass die Silanmoleküle an die Gipsoberfläche adsorbieren, um einen hydrophobierenden Effekt zu erzeugen. Die...
Die Simulationen basierten auf der Annahme, dass die Silanmoleküle an die Gipsoberfläche adsorbieren, um einen hydrophobierenden Effekt zu erzeugen. Die Adsorptions-/Desorptionsvorgänge wurden einzeln für jedes der verschiedenen möglichen Hydrolyse- und Kondensationsprodukte (Reaktionen 1-6, Teil 1 in ZKG 7-8/2013) des Propyltriethoxysilans simuliert. Entsprechend der Protolyse von Kieselsäure (pKS1 = 9,51, pKS2 = 11,74) [26] wurde die Bildung von bis zu zwei freien Silanol-Gruppen durch die Hydrolyse der Ethoxy-Gruppen bei einem pH-Wert von 12,5 angenommen. Zu den verschiedenen hydrolisierten Spezies wurden auch die dimeren und trimeren Formen von Propyltriethoxysilan für die Simulationen herangezogen (Tab. 2).
Die Simulationen wurden im kanonischen (NVT-) Ensemble unter periodischen Rahmenbedingungen sowie unter Verwendung des Berendsen-Thermostats durchgeführt, um eine Temperatur von 298 K aufrecht zu erhalten. Ein Cutoff von 14 Å wurde für die van-der-Waals-Wechselwirkungen angesetzt. Coulombsche Wechselwirkungen wurden mittels Particle-Mesh-Ewald-Summe (PME) mit einem Impulsraum-Cutoff von 14 Å berechnet. Für die Integration der Bewegungsgleichung wurde der Leapfrog-Algorithmus in Zeitschritten von jeweils 1 Femtosekunde (1fs = 10‑15 s) verwendet.
Da die Silanmoleküle mit allen Gipskristallflächen wechselwirken sollten, um eine hydrophobierende Wirkung hervorzurufen, wurde beispielhaft aber nur die häufig auftretende Kristallfläche (020) betrachtet. Für die Simulationen wurde ein 15 Å-dickes Atommodel (Gipskristallstruktur nach Pedersen und Semmingsen [27]) mit Schnitt entlang der (020) Kristallfläche erzeugt. Um die Kristallstruktur exakt reproduzieren zu können, wurde eine Simulationsbox (x = 52,2 Å, y = 37,7 Å, z = 80,2 Å, b = 127,4°) verwendet. Alle Atome der Randschicht wurden in ihren Positionen während der Simulationen fixiert. Oberhalb der Randschicht wurde eine 65 Å dicke Vakuumschicht erzeugt. Die Silanmoleküle wurden in den Vakuumbereich eingebracht. Simulationen wurden sowohl mit als auch ohne Wasser als Lösungsmittel durchgeführt. Für die Lösungsmittelsimulationen wurde der restliche Vakuumbereich mit „Simple Point Charge“ (SPC, einfache Punktladung) Wasser gefüllt [28].
Um ohne Annahmen die günstigsten Positionen und Konformationen der Silanmoleküle auf der Gipsoberfläche zu erhalten, wurde das zu untersuchende Silanmolekül in einer zufälligen Position und Konformation in der Vakuumschicht oberhalb der Oberfläche positioniert, wonach 100 Pikosekunden (ps) zum Erreichen der Gleichgewichtslage zugelassen wurden. Dieser Vorgang wurde 50 mal für jede Silanspezies wiederholt. Diese Simula-tionen mussten ohne Lösungsmittel durchgeführt werden, da Wassermoleküle eine ausgeprägte Hydratationsschicht auf der Gipsoberfläche bilden, so dass die Adsorption eines Moleküls aus der freien Lösung an die Gipsoberfläche kinetisch stark gehemmt ist. Die Simulationszeit des Adsorptionsvorgangs in Anwesenheit von Wasser wird somit unpraktikabel lang. Ohne Lösungsmittel erreichen die Silanmoleküle die Oberfläche binnen einiger ps. Die so erhaltenen Konformationen der adsorbierten Silane an der Oberfläche wurden verglichen und die energetisch günstigsten als Ausgangskonformationen für weitere Simulationen ausgewählt.
Die Stärke der Wechselwirkung zwischen dem Silanmolekül und der Gipsoberfläche wird durch die Wechselwirkungsenergie ausgedrückt. Je negativer die Wechselwirkungsenergie, desto stärker die Adsorption. Die Wechselwirkungsenergie Eint wird anhand folgender Gleichung berechnet: Eint = Etot - Esurf - Emol (Gleichung 1).
Darin ist Etot die Energie der gesamten Simulationsbox, bestehend aus der Gipsoberfläche und des adsorbierten Silanmoleküls und wird als Mittelwert von fünf einzelnen MD-Simulationen von jeweils 100 ps Dauer ausgegeben. Esurf ist die Energie der reinen Oberfläche ohne jegliche Silanmoleküle (Esurf = -772649,2 kJ/mol). Emol gibt die Energie des reinen Silanmoleküls ohne Oberfläche wieder und wird als Mittelwert einer MD-Simulation von 100 ps Dauer unter Vakuum erhalten.
Da das Lösungsmittel Wasser einen entscheidenden Einfluss auf die Adsorption hat, wurden auch entsprechende Simulationen in Gegenwart von Wasser durchgeführt. Wie schon erwähnt, konnte die Simulation des Adsorptionsvorgangs in Anwesenheit von Wasser nicht realisiert werden. Möglich war jedoch die Simulation der Wechselwirkung zwischen Silan und Oberfläche über den Desorptionsvorgang, ausgehend von den über Vakuumsimulationen vorgefundenen adsorbierten Konformationen.
Dazu wurden die energetisch günstigsten adsorbierten Konformationen der verschiedenen Silanspezies auf der Gipsoberfläche solvatisiert und die so erhaltenen Simulationsboxen in drei aufeinanderfolgenden Schritten äquilibriert. Während des ersten Schrittes konnte das Wasser äquilibrieren, während die Atomlagen der Oberfläche und der Silanatome fixiert blieben. Mit dem zweiten Schritt wurden die Silanmoleküle innerhalb der neuen wasserhaltigen Umgebung äquilibriert. Im dritten Schritt erfolgte das Äquilibrieren von Wasser und Silanmolekülen gemeinsam.
Sobald sich Lösungsmittelmoleküle in der Simulationsbox befinden, eignet sich die Wechselwirkungsenergie des Systems zur Beurteilung der Wechselwirkungsstärke wegen der erheblichen statistischen Schwankungen nicht mehr. Stattdessen wurde daher das Potential der mittleren Kraft (engl. potential of mean force, PMF) verwendet, welches mittels Pullcode erhalten wurde. Bei diesem Vorgang wurde der Massenmittelpunkt (engl. centre of mass, COM) der adsorbierten Moleküle in jedem Zeitschritt einen definierten, schwindend kleinen Wegbetrag von der Oberfläche entfernt. Alle anderen Atome konnten sich frei bewegen. Der Abstand zwischen dem COM und der Oberfläche wird mithilfe einer zurückhaltenden Kraft aufrechterhalten. Durch Integration dieser haltenden Kraft (PMF) über den Abstand wird die freie Desorptionsenergie ∆Fdesorb erhalten. Je positiver diese ist, desto stärker die Wechselwirkung zwischen dem Silanmolekül und der Oberfläche. COM-Pulling-Simulationen wurden für alle monomeren Silanspezies durchgeführt, jeweils beginnend bei den fünf unterschiedlichen Konformationen (nach den Momentaufnahmen der MD-Simulationen von Schritt 2 und 3 der Äquilibrierung der adsorbierten Silanspezies in Gegenwart von Wasser).
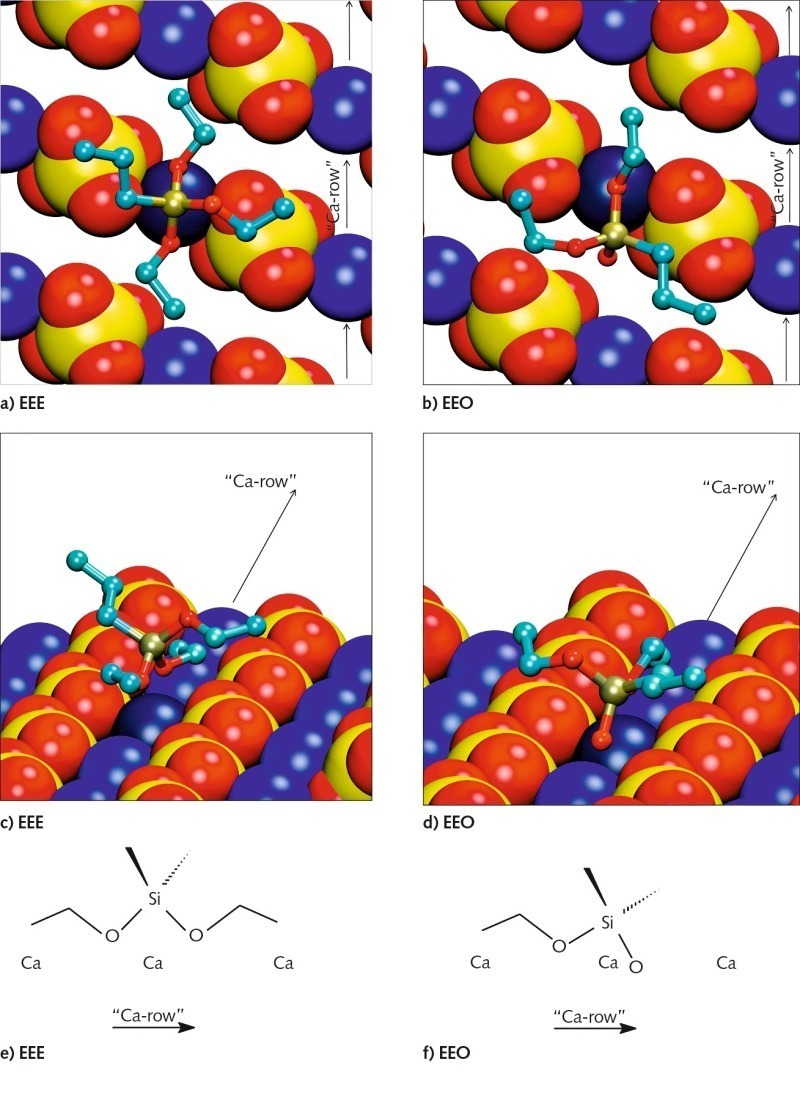
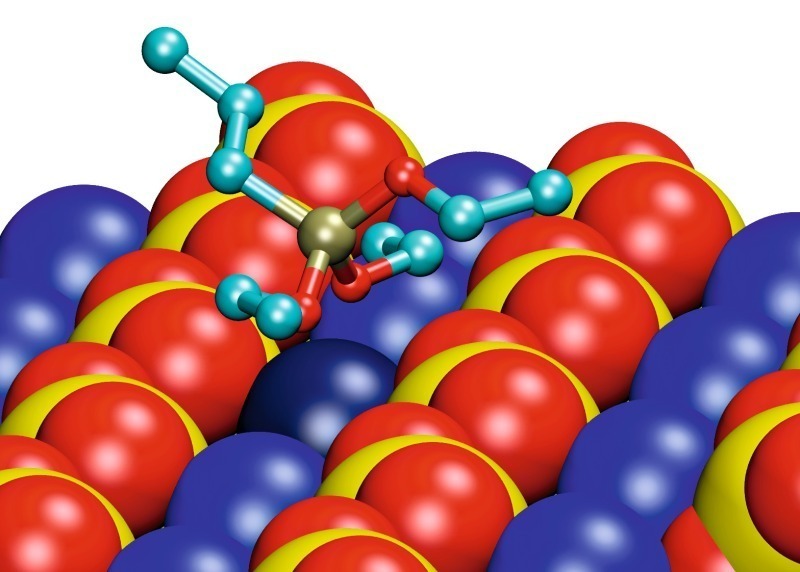
Die energetisch günstigsten Konformationen der an die Gipsoberfläche adsorbierten monomeren Silane sind für die Beispiele Propyltriethoxysilan (EEE) und Propyldialkoxysilanol (EEO) in Bild 7 gezeigt. Die Konformationen der unterschiedlichen Silanmoleküle zeigen einige Gemeinsamkeiten. Es gibt Reihen von Calciumionen zwischen den Reihen von Sulfationen langs der (020)-Gips-oberflache. Das Siliciumatom im Silanmolekül ist tetra-edrisch koordiniert. Die Wechselwirkung zwischen dem Silan und der Oberfläche wird von den Sauerstoffatomen des Silans bestimmt, die die Calciumionen der Oberfläche koordinieren. Zwei der drei Ethoxy-Gruppen des Silans sind zur Oberfläche längs des Calciumionenkanals orientiert und koordinieren dasselbe Calciumion (dunkelblau in Bild 7). Die dritte Ethoxy-Gruppe sowie die Propyl-Gruppe zeigen von der Gipsoberfläche weg.
Die Wechselwirkung zwischen dem Sauerstoffatom und dem Calciumion wird stärker und der interatomare Abstand geringer mit abnehmender sterischer Hinderung und zunehmender Ladung des Sauerstoffatoms. Daher nimmt die Wechselwirkung ausgehend vom Sauerstoff in Ethoxy-Gruppen, über Sauerstoff in der Hydroxyl-Gruppe, hin zu freiem Sauerstoff zu. Aus diesem Grund nähert sich das Siliciumatom der Oberfläche in der Reihenfolge EEE über EEO bis EOO (Propylethoxysilandiol) zunehmend. Da maximal zwei freie Sauerstoffatome mit der Oberfläche wechselwirken können, wurden ähnliche Konformationen für EOO und HOO (Propylsilantriol) gefunden. Es spielt also keine Rolle, ob die dritte Ethoxy-Gruppe hydrolysiert ist oder nicht.
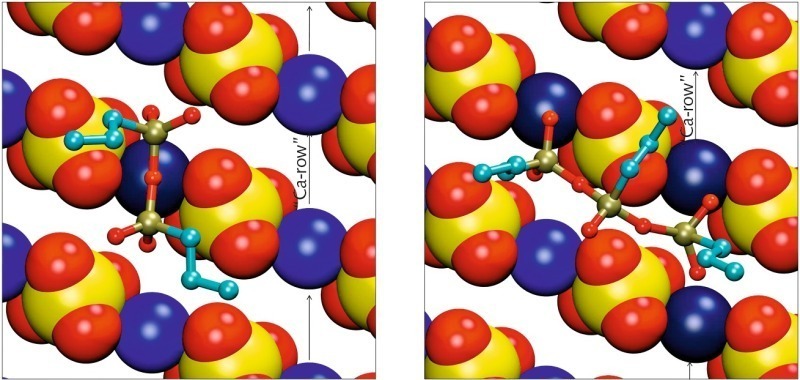
Die Situationen für die dimeren und trimeren Formen des PTES zeigt Bild 8. Das Dimer kann aus sterischen Gründen nur mit zwei seiner vier freien endständigen Sauerstoffatome adsorbieren. Die anderen beiden freien Sauerstoffatome sowie das Sauerstoffbrückenatom zwischen den beiden Siliciumatomen zeigen von der Oberfläche weg. Während das dimere Silan längs des Calciumkanals ähnlich wie die monomeren Silane ausgerichtet ist, koordiniert die trimere Form eher Calciumionen in zwei benachbarten Kanälen. Die beiden endständigen Siliciumatome adsorbieren jeweils über zwei freie Sauerstoffatome, während das mittige Silicumatom zusammen mit seinem freien Sauerstoffatom von der Oberfläche weg zeigt und den Sulfatkanal zwischen den beiden Calciumkanälen überbrückt.
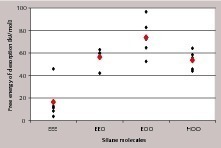
Die in Bild 9 angegebenen Wechselwirkungsenergien sind so leicht verständlich. Mit zunehmender Anzahl der freien Sauerstoffatome je Siliciumatom nimmt die Wechselwirkung in der Reihenfolge EEE, EEO bis hin zu EOO zu. Ob die dritte Ethoxy-Gruppe hydrolysiert vorliegt oder nicht, hat keinen Einfluss auf die Wechselwirkungsenergie (in Abwesenheit des Lösungsmittels), da EOO und HOO annähernd die gleichen Werte aufweisen. Im Fall der dimeren Form steht pro Siliciumatom nur ein freies Sauerstoffatom in Wechselwirkung mit der Oberfläche, was zu Wechselwirkungsenergien ähnlich denen von EEO führt. Im Falle der trimeren Form steigt die sterische Hinderung weiter, was eine weitere geringere Wechselwirkung je Siliciumatom zur Folge hat.
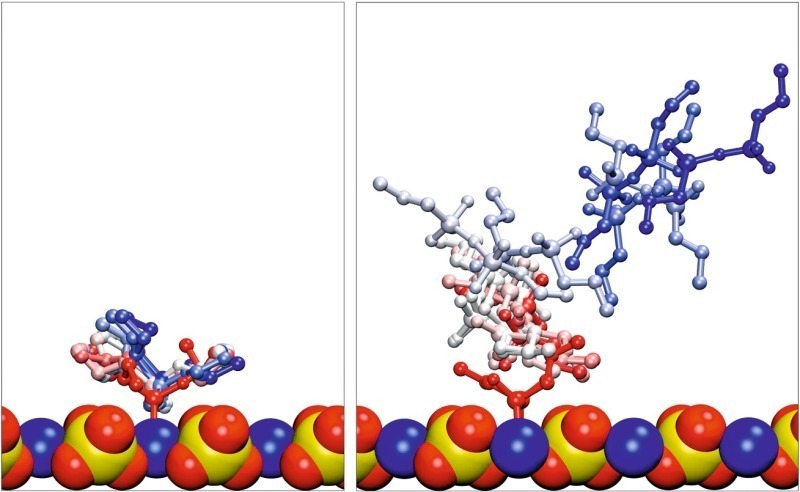
Um bisher vernachlässigte Lösungsmitteleffekte zu berücksichtigen, wurden die adsorbierten Konformationen der verschiedenen Silanmoleküle solvatisiert. Während der aufeinanderfolgenden Äquilibrierungsschritte blieben die monomeren Silane mit nur einer geringfügigen Zunahme des Abstands zur Oberfläche adsorbiert, während die dimeren und trimeren Formen in jeder Simulation desorbierten, wie Bild 10 zeigt. Es ist die Überlagerung von Schnappschüssen der Simulation von EOO beispielhaft für monomere Silane und für D4O (Dimer) als Beispiel der Kondensatformen gezeigt. Daraus ergibt sich, dass bei Anwesenheit des Lösungsmittels die Wechselwirkung zwischen den Kondensationsprodukten von PTES und der Gipsoberfläche wesentlich geringer ist als im Falle der monomeren Formen. Die dimere und trimere Formen wurden deshalb nicht weiter betrachtet.
Um die Wechselwirkungsstärke der verschiedenen monomeren Silanspezies in Anwesenheit des Lösungsmittels zu beurteilen, wurde das PMF berechnet und integriert, um die freie Desorptionsenergie zu erhalten. Je positiver die freie Desorptionsenergie, desto stärker ist die Adsorption des Silanmoleküls. Bild 11 zeigt die erhaltenen Einzel- und Mittelwerte der freien Desorptionsenergie für die verschiedenen Silane. Die stärkste Wechselwirkung mit der Gipsoberfläche ergibt sich wieder für das zweifach hydrolisierte monomere Propylethoxysilandiols (EOO), gefolgt von EEO (einfach hydrolisiertes Monomer) und HOO (vollkommen hydrolisiertes Monomer).
4 Zusammenfassung
Bei einer Hydrolysezeit > 35 min ist die hydrophobierende Wirkung rückläufig. 29Si-NMR-spektroskopische Untersuchungen zeigen, dass vornehmlich Propyldiethoxysilanol (EEO), eines der drei möglichen Produkte der PTES-Hydrolyse, in diesem Zeitfenster gebildet wird. Anschließend, zwischen 30 und 45 min, wurden Kondensationsprodukte durch 29Si-NMR-Spektroskopie nachgewiesen [Resultate aus Teil 1, ZKG 7-8/2013, S. 72-78]. Molekulardynamische (MD) Simulationen bestätigten, dass die Hydrophobierung von Gipsbaustoffen durch Alkylalkoxysilane auf der Wechselwirkung von hydrolisierten PTES-Spezies mit der Gipsmatrix beruht. Die stärkste Wechselwirkung mit Gipskristallflächen zeigt das zweifach hydrolisierte monomere Propylethoxysilandiol (EOO). Die zweitstärkste Wechselwirkung wird mit dem einfach hydrolisierten monomeren Propyldiethoxysilanol (EEO) erreicht. Die MD-Simulationen zeigen, dass die Kondensationsprodukte keine signifikanten Wechselwirkungen mit der Gipskristallfläche aufweisen. Die Ergebnisse von Hydrolyseversuchen mit einer längeren Vorhydrolysezeit werden somit bestätigt. Die Hydrophobierung von Gipsbaustoffen durch Alkylalkoxysilane hängt demzufolge vom Grad der Hydrolyse des Alkyltriethoxysilans, also von den monomeren Hydrolyseprodukten ab. Für die Wirkung müssen die Hydrolyseprodukte innerhalb der Abbindezeit des Gipsproduktes gebildet werden. Die Hydrolysereaktion läuft mit einer entsprechenden Geschwindigkeit sowohl im sauren als auch im basischen pH-Bereich ab, im neutralen pH-Bereich dagegen sehr langsam. Für Anwendungen von Gipsprodukten kommt nur der basische pH-Wertbereich in Betracht. Dem allgemeinen Verarbeitungszeitraum von Gipsprodukten entsprechend, liegt für Alkyltriethoxysilane mit n < 5 die zur Bildung durch Hydrolyse von monomeren Silanspezies benötigte Zeit zum Erreichen einer hydrophobierenden Wirkung im entsprechenden Zeitfenster. Die Hydrolysezeit der längerkettigen Alkyltriethoxysilane (n > 5) ist, wie oben für den Fall des Octyltriethoxysilans (n = 8) gezeigt, zu lang, um einen hydrophobierenden Effekt zu erzielen. Bei neutralem pH-Wert weisen selbst kurzkettige Alkylalkoxysilane keine hydrophobierende Wirkung auf, da auch hier die Hydrolysezeit zur Bildung von wirksamen Silanolmolekülen wieder zu lang ist. Mit der Verwendung eines effizienteren Katalysators (wie z.B. organometallische Katalysatoren) könnten einige Alkylalkoxysilane auch außerhalb eines hohen oder niedrigen pH-Wertes funktionieren, wie im Patent [4] beschrieben wird.
5 Danksagung
tab ZKG KOMBI Test
Es handelt sich hierbei um ein Testangebot. Es berechtigt zu keinem gültigen Abonnement und steht hier rein für Testläufe. Bitte diesem Prozess nicht folgen.
Es handelt sich hierbei um ein Testangebot. Es berechtigt zu keinem gültigen Abonnement und steht hier rein für Testläufe. Bitte diesem Prozess nicht folgen.
tab ZKG KOMBI Study Test
Es handelt sich hierbei um ein Testangebot. Es berechtigt zu keinem gültigen Abonnement und steht hier rein für Testläufe. Bitte diesem Prozess nicht folgen.
Es handelt sich hierbei um ein Testangebot. Es berechtigt zu keinem gültigen Abonnement und steht hier rein für Testläufe. Bitte diesem Prozess nicht folgen.