Hydrophobizing of gypsum by silanes (Part 2)

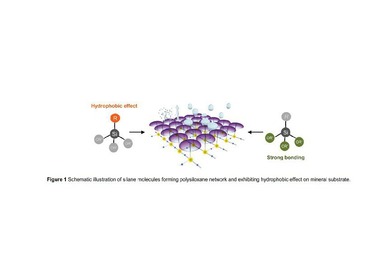
The effectiveness of silanes as known water repellents for gypsum building materials had been the topic of the experimental studies on the example of propyltriethoxysilane in Part 1 of this article (ZKG 7-8/2013, pp. 72-78). Part 2 considers the involved processes on a molecular level, using molecular dynamic simulations. The results are interpreted together with the findings of Part 1 to obtain an understanding of the working mechanism of the hydrophobizing effect of silanes, especially alkyltriethoxysilanes, using the example of propyltriethoxysilane.
The simulations were based on the assumption that the silane molecules adsorb on the gypsum surface to achieve the hydrophobizing effect. The adsorption/desorption processes were...
The simulations were based on the assumption that the silane molecules adsorb on the gypsum surface to achieve the hydrophobizing effect. The adsorption/desorption processes were separately simulated for each of the different possible hydrolysis and condensation products (reactions 1–6, Part 1 in ZKG 7-8/2013) of propyltriethoxysilane. According to the protolysis of silicic acid (pKS1 = 9.51, pKS2 = 11.74) [26] it can be assumed that up to two free silanol groups formed by hydrolysis of the ethoxy groups will be deprotonated at a solution pH of 12.5. In addition to the different hydrolyzed species the dimeric and trimeric forms of propyltriethoxysilane were used for the simulations (Tab. 2).
The simulations were performed in the NVT ensemble with periodic boundary conditions and the Berendsen thermostat to keep the temperature at 298 K. A cut-off of 14 Å was used for van der Waals interactions. Coulomb interactions were calculated by means of the Particle-Mesh Ewald summation (PME) with a real-space cut-off of 14 Å. The integration of the equation of motion was done by the leapfrog algorithm with a time step of 1 femtosecond (1fs = 10–15 s).
Since the silane molecules must interact with all gypsum surfaces to evoke a hydrophobizing effect, only the main occurring gypsum surface, (020), was examined as an example. For the simulations a 15 Å thick atomistic model of the cleaved (020) surface of gypsum was generated based on the experimental crystal structure from Pedersen and Semmingsen [27]. To exactly reproduce the crystal structure a simulation box was used (x = 52.2 Å, y = 37.7 Å, z = 80.2 Å, b = 127.4°). All atoms in the surface layer were fixed in their positions during the simulations. Above the surface layer a 65 Å thick vacuum layer was installed. Silane molecules were placed in the vacuum region. Simulations were carried out with and without water as solvent. For the preparation of solvent simulations the remaining vacuum region was filled with Simple Point Charge (SPC) Water [28].
To get the most favorable positions and conformations of the silane molecules on the gypsum surface without any assumptions, the silane molecule under investigation was placed at random position and conformation in the vacuum slab above the surface and allowed to equilibrate for 100 picoseconds (ps). This procedure was repeated 50 times for every silane species. These simulations had to be carried out without solvent, because water molecules form a strong hydration layer on the gypsum surface, so that the adsorption of a molecule from free solution onto the surface is strongly kinetically hindered. That makes the simulation of the adsorption process in the presence of water impractical regarding the necessary simulation time. Without solvent the silane molecules reach the surface within a few ps. The obtained conformations of the adsorbed silanes on the surface were compared and the energetically most favorable ones were chosen as starting conformations for further simulations.
The strength of the interaction between silane mole-cule and gypsum surface is expressed in terms of the interaction energy. The more negative the interaction energy is, the stronger is the adsorption. The interaction energy, Eint, is calculated with the following equation: Eint = Etot - Esurf - Emol (eq. 1).
Therein Etot is the energy of the total simulation box consisting of the gypsum surface and the adsorbed silane molecule. Etot is obtained as the average of five separate 100 ps MD simulations. Esurf is the energy of the pure surface without any silane molecules (Esurf = -772649.2 kJ/mol). Emol is the energy of the pure silane molecule without surface. It is obtained as the average of a 100 ps MD simulation in vacuum.
Normally, the solvent water has a decisive influence on the adsorption. To account for the solvent effects, simulations with solvent were also carried out. Since the simulation of the adsorption process was impractical in the presence of water, the interaction between silane and surface was investigated by means of the desorption process instead, starting from the adsorbed conformations found during the vacuum simulations.
Therefore, the energetically most favorable adsorbed conformations of the different silane species on the gypsum surface found in vacuum simulations were solvated and the obtained simulation boxes equilibrated in three steps. During the first step the water was allowed to equilibrate, keeping the atomic positions of surface and silane atoms fixed. During the second step the silane molecule was allowed to equilibrate within the new water environment, and during the third step water and silane molecules were equilibrated together.
With solvent molecules within the simulation box, the interaction energy of the system becomes inappropriate for the assessment of the interaction strength because of the large statistical fluctuations. The potential of mean force (PMF) was used instead. The PMF was obtained using the pull code. In doing so, the center of mass (COM) of the adsorbed molecules was moved a well defined, infinitesimal small distance away from the surface each time step, apart from that allowing all atoms to move freely. The distance between the COM and the surface is maintained with the help of a constraint force. By integrating this constraint force (PMF) over the distance, the free energy of desorption ∆Fdesorb is obtained. The more positive the free energy of desorption is, the stronger is the interaction between silane molecule and surface. COM-pulling simulations were carried out for all monomeric silane species starting from five different conformations each (after the snapshot of the second and third equilibration step from the MD simulations of the adsorbed silanes with solvent).
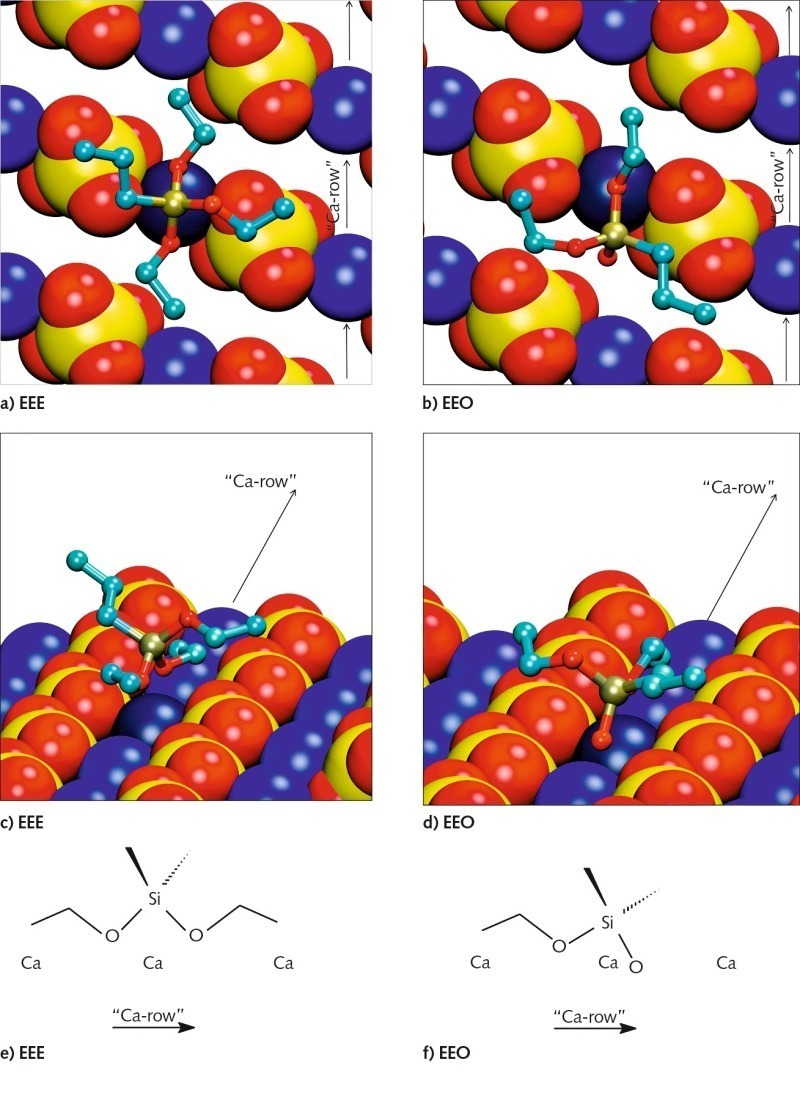
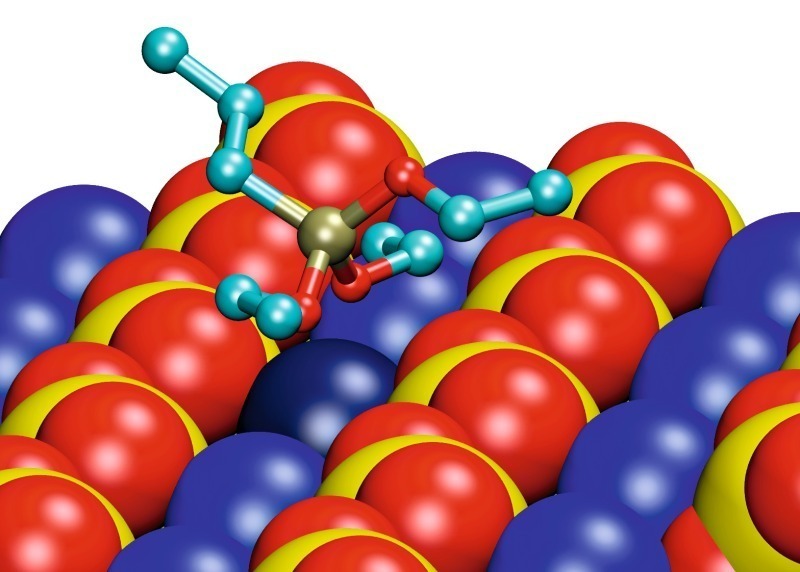
The energetically most favorable conformations of the adsorbed monomeric silanes at the gypsum surface are demonstrated in Figure 7 for the examples of propyltriethoxysilan (EEE) and propyldialkoxysilanol (EEO). The conformations of different silane molecules have some common aspects. There are calcium ion rows between sulfate ion rows along the (020) gypsum surface. The silicon atom in the silane molecule is tetrahedrally coordinated. The interaction between silane and surface is governed by the oxygen atoms of the silane, coordinating the calcium ions of the surface. Two of the three ethoxy groups of the silane are oriented towards the surface along the calcium ion channel, coordinating the same calcium ion (marked in darker blue in Fig. 7).The third ethoxy group and the propyl group are oriented away from the gypsum surface.
The interaction between the oxygen atom and the calcium ion is getting stronger and the interatomic distance is getting smaller, with decreasing sterical hindrance and increasing atomic charge of the oxygen atom. Thus, the interaction increases in order from oxygen in ethoxy groups, via oxygen in hydroxyl group to free oxygen. That is why the silicon atom gets closer to the surface in the order from EEE via EEO to EOO (propylethoxysilandiol). Since a maximum of two free oxygen atoms can interact with the surface, similar conformations were found for EOO and HOO (propyl-silantriol). Thus, it is irrelevant whether or not the third ethoxy group is hydrolyzed.
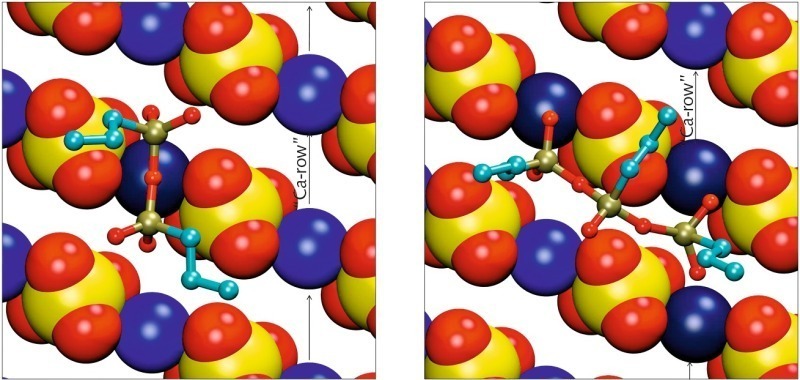
For the dimeric and trimeric form of PTES the situation is shown in Figure 8. For sterical reasons the dimer can only adsorb with two of its four terminating free oxygens. The other two free oxygens as well as the bridging oxygen between the two silicon atoms are oriented away from the surface. While the dimeric silane is aligned along the calcium channel similar to the monomeric silanes, the trimeric form coordinates calcium ions in two neighboring channels. The two terminating silicon atoms adsorb via two free oxygen atoms each, the central silicon atom together with its free oxygen is oriented away from the surface, bridging the sulfate channel between the two calcium channels.
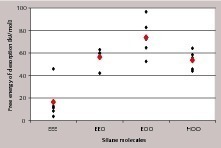
From this the interaction energies plotted in Figure 9 can easily be understood. With an increasing number of free oxygen atoms per silicon atom the interaction increases in the order EEE, EEO to EOO, expressed by more negative values of the interaction energy. Whether or not the third ethoxy group is hydrolyzed does not have any influence on the interaction energy (in the absence of solvent), as EOO and HOO show nearly the same values. For the dimeric form there is one free oxygen atom interacting with the surface per silicon atom only, leading to interaction energies similar to that of EEO. For the trimeric form there is further increase of steric hindrance and thus less interaction per silicon atom.
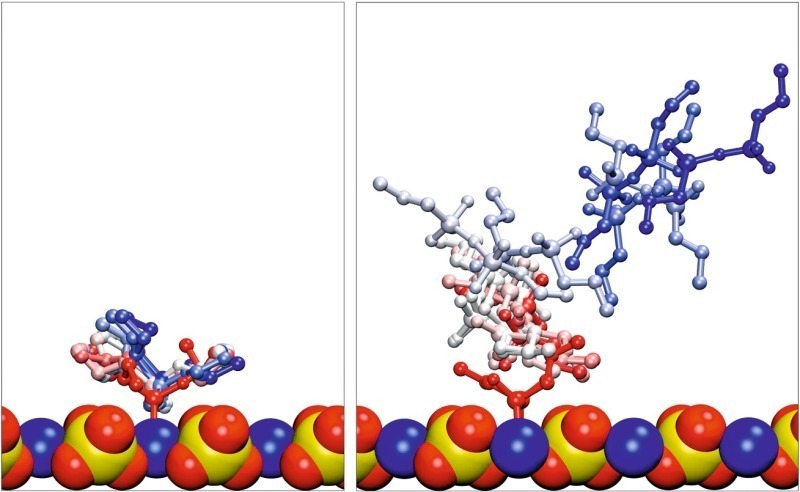
To account for solvent effects not considered until now, the adsorbed conformations of the different silane molecules were solvated. During the following equilibration steps, the monomeric silane forms remained adsorbed, only slightly increasing the distance to the surface, whereas the dimeric and trimeric forms desorbed in every simulation, which is illustrated in Figure 10. It shows multiple views for the simulation of EOO as example for monomeric silanes and D4O as an example of the condensate forms. Thus, it can be concluded, that in the presence of solvent the interaction between the condensates of PTES and the gypsum surface is considerably smaller than for the monomeric forms. Therefore, the dimeric and trimeric forms were not considered any further.
To access the interaction strength of the different monomeric silane species in the presence of solvent, the potential of mean force was calculated and integrated to obtain the free energy of desorption. The more positive the free energy of desorption is, the stronger is the adsorption of the silane molecule. The obtained single and average values of the free energy of desorption for the different silanes are shown in Figure 11. The strongest interaction with the gypsum surface results for the twice hydrolyzed monomer propylethoxysilandiol (EOO) again, followed by EEO (single hydrolyzed monomer) and HOO (complete hydrolyzed monomer).
4 Conclusion
For a hydrolysis time > 35 minutes the hydrophobization effect decreases again. 29Si-NMR-spectroscopic investigations reveal that mainly propyldiethoxysilanol (EEO), one of the three possible hydrolysis products of PTES, is formed within this time frame. In the time scale between 30 and 45 minutes condensation products were detected by 29Si-NMR [Results from Part 1, ZKG 7-8/2013, pp. 72-78]. Molecular dynamic (MD) simulations could verify that the effect of reduced water uptake of gypsum products using alkylalkoxysilanes is due to the interaction of hydrolyzed PTES species with the gypsum matrix. Thereby, the strongest interaction with the gypsum crystal faces was formed with the twice hydrolyzed monomer propylethoxysilandiol (EOO). For the single hydrolyzed monomer propyldiethoxysilanol (EEO) the second strongest interaction was formed. According to the MD simulations the condensation products show no significant interactions with the gypsum crystal face. This is in line with the results of hydrolysis experiments with prolonged pre-hydrolysis time (Part 1). As a conclusion, hydrophobization of gypsum based products by alkylalkoxysilanes depends on the degree of alkyltriethoxysilane hydrolysis and consequently on the monomeric hydrolysis products. In order to be effective, hydrolysis products have to be formed within the setting time of the gypsum product. The hydrolysis reaction has an appropriate rate in the acidic as well as alkaline pH value range, and is very low in the neutral range. For gypsum applications only the alkaline pH value comes into consideration. Consistent with the common workability time of gypsum products, for alkyltriethoxysilanes with n < 5 the time scale to form monomeric silane species by hydrolysis is in the right order to cause a hydrophobization effect. For longer chain alkyltriethoxysilanes (n > 5) the hydrolysis time becomes too long as shown for octyltriethoxysilane (n = 8), resulting in a water uptake in the same order of magnitude as without hydrophobizing additive. At neutral pH also short chain alkylalkoxysilanes no longer cause a hydrophobization effect of gypsum products as again the hydrolysis time to form the effective silanol molecules becomes too long. By using a more efficient catalyst than high or low pH (e.g. organo metal catalysts), some alkylalkoxysilanes might work as described in the patent [4].
5 Acknowledgement
Überschrift Bezahlschranke (EN)
tab ZKG KOMBI EN
This is a trial offer for programming testing only. It does not entitle you to a valid subscription and is intended purely for testing purposes. Please do not follow this process.
This is a trial offer for programming testing only. It does not entitle you to a valid subscription and is intended purely for testing purposes. Please do not follow this process.
tab ZKG KOMBI Study test
This is a trial offer for programming testing only. It does not entitle you to a valid subscription and is intended purely for testing purposes. Please do not follow this process.
This is a trial offer for programming testing only. It does not entitle you to a valid subscription and is intended purely for testing purposes. Please do not follow this process.